 2025-03-27
2025-03-27
 分享至:
分享至:
近日,我校物理学院韩永昌教授课题组与中国科学院大连化学物理研究所化学反应动力学全国重点实验室傅碧娜研究员、张东辉院士团队合作,在《自然》子刊《自然-通讯》(Nature Communications)上发表题为“Cl + C2H2→ C2H + HCl双分子反应中的专属漫游机制”(Exclusive roaming mechanism for the Cl+C2H2→ C2H+ HCl bimolecular reaction)的论文。我校博士生白玉瑶为共同第一作者,大工为第一单位。
氯原子与乙炔碰撞反应不仅是乙炔在海洋和极地对流层的主要去除途径,也是平流层下部卤素原子的重要消耗途径。深入研究其内在反应机制,有助于理解大气卤素化学循环的关键过程,为气候模型提供更精确的反应动力学参数,同时为评估人为排放卤代烃对臭氧层的影响提供重要理论依据。

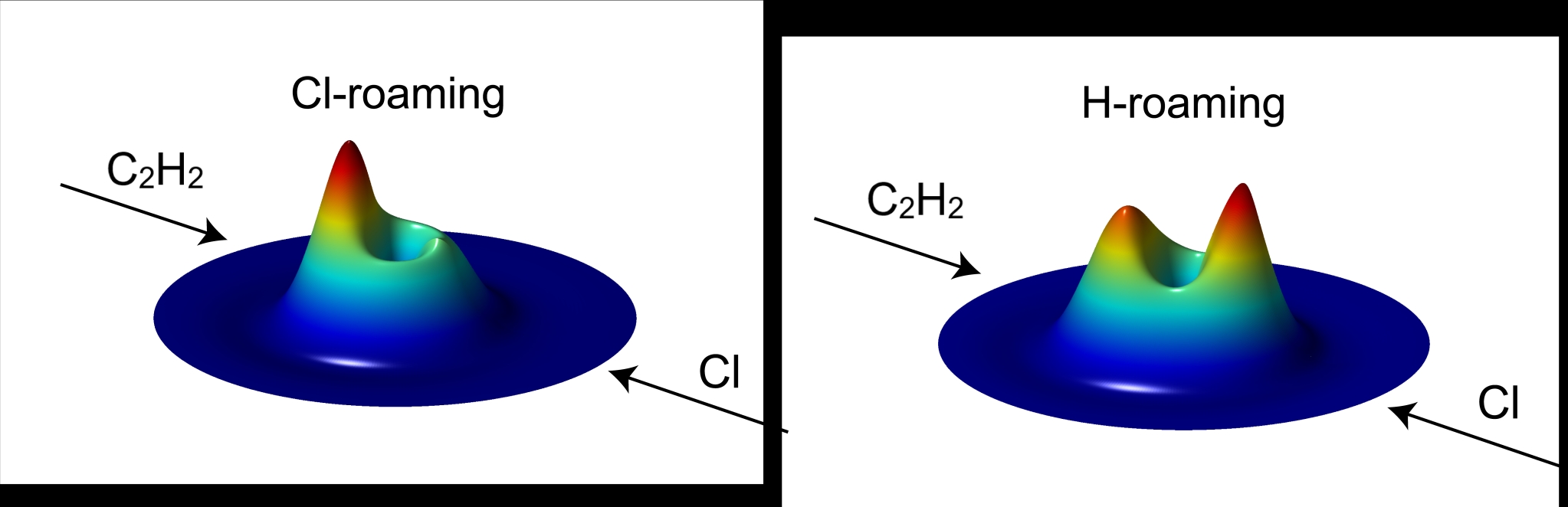
Cl + C2H2体系势能面形貌图
研究人员通过构建高精度全维基态势能面,并在此基础上对Cl+C2H2→HCl+C2H反应开展了可靠的动力学计算工作。研究结果表明,在该碰撞反应中除经传统过渡态结构的直接抽取路径,还存在氯漫游(Cl-roaming)和氢漫游(H-roaming)两种漫游机制。在氯漫游机制中,入射的氯原子与乙炔分子碰撞后会形成络合物中间体(C2H2Cl),随后氯原子从络合物中部分解离并在乙炔分子附近漫游,最终抽取其上氢原子形成氯化氢分子。而在氢漫游机制中,氢原子率先从形成的络合物中间体中脱离,并在络合物附近迁移漫游,进而从中提取Cl原子生成产物。
漫游路径的产物平动能和散射角分布的3D极坐标图
通过进一步分析其动力学计算结果,研究发现漫游机制在该吸热反应中的贡献几乎为100%,直接抽取路径与之相比几乎可以忽略不计。这些发现挑战了传统过渡态理论对双分子反应的认知,强调了在反应动力学研究中考虑非传统路径对精确预测速率常数和深入理解反应机理的重要性。

不同反应路径的积分截面
近年来,韩永昌在国家自然科学基金等项目的支持下,在分子碰撞漫游及能量转移理论研究方面取得了持续性的突出进展,相关成果发表于《化学科学》(Chemical Science)、《物理化学快报》(The Journal of Physical Chemistry Letters)以及《自然-通讯》(Nature Communications)等权威期刊。
来源:物理学院
编辑:常思萌
审核:赫铭 王增强
 最新动态
最新动态
 投稿入口
投稿入口
 使用说明
使用说明